Short Answer
Definition of Heme and Its Molecular Structure
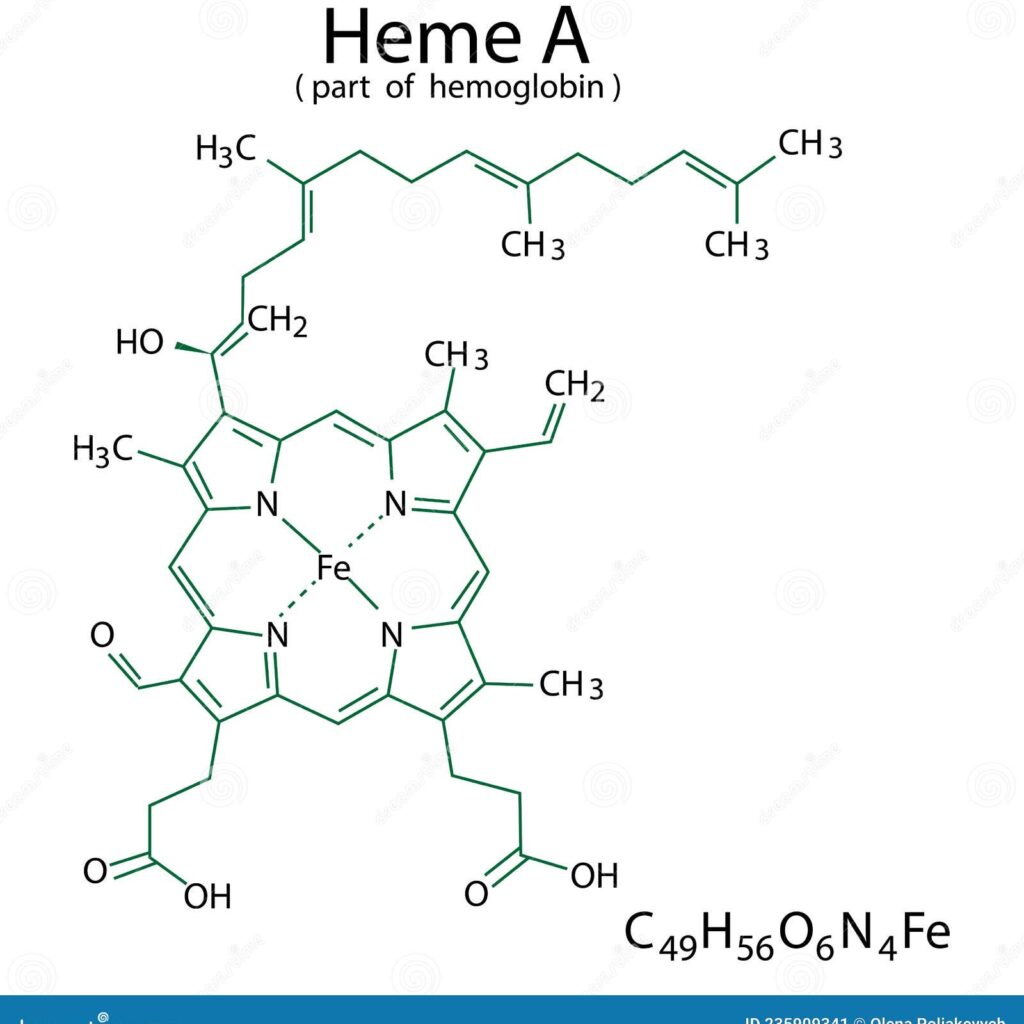
Heme is a vital organic molecule characterized by a complex molecular framework that plays essential roles in various biological functions. It consists of a central iron (Fe) atom embedded within a planar, cyclic tetrapyrrole known as a porphyrin ring. This structure is fundamental to the function of hemoproteins such as hemoglobin and myoglobin, as well as numerous enzymes. The porphyrin ring acts as a chromophore, responsible for absorbing light and imparting the distinctive red color to blood.
Molecular Orbital Theory Applied to Heme
Molecular Orbital (MO) theory offers a comprehensive approach to understanding how atomic orbitals combine to form molecular orbitals, which govern the electronic properties and reactivity of molecules like heme. In heme, the atomic orbitals from carbon, nitrogen, and iron atoms merge to create unique molecular orbitals. These include the π (pi) and σ (sigma) orbitals, which are crucial for bonding interactions and electron distribution within the molecule.
Role of π Orbitals in the Porphyrin Ring
The π orbitals arise from the conjugated system of the porphyrin ring, where delocalized electrons are shared across the four nitrogen-containing pyrrole units. This delocalization generates a series of molecular orbitals, including bonding (lower-energy) and antibonding (higher-energy) types. Of particular importance are the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO), as their energy levels and spatial arrangements influence the electronic transitions that underpin heme’s biochemical functions.
Iron’s d Orbitals and Their Interactions
In its fully reduced form, the iron atom in heme exhibits a d⁶ electronic configuration. The interaction between the iron’s d orbitals and the p orbitals of the surrounding nitrogen atoms is central to the molecule’s electronic structure. Upon oxygen binding, the iron undergoes an electronic rearrangement involving π-backbonding, where electron density is shared between the filled d orbitals of iron and the vacant π* orbitals of molecular oxygen (O₂). This interaction stabilizes the heme-oxygen complex and enhances oxygen affinity, facilitating cooperative binding in hemoglobin.
Geometric and Electronic Influences on Heme Functionality
The planar geometry of heme supports sp² hybridization of the carbon atoms within the porphyrin ring, which complements the delocalized π electron system and maintains the molecule’s rigid structure. Changes in this geometry, such as those induced by axial ligands like nitric oxide or carbon monoxide, alter the p-orbital interactions and significantly impact the electronic properties and reactivity of heme. These structural modifications highlight heme’s adaptability in various biological reactions and signaling pathways.
Impact of Substituents on the Porphyrin Core
Substituent groups attached to the periphery of the porphyrin ring can profoundly affect the molecular orbital landscape of heme. These groups influence the electron-donating or electron-withdrawing characteristics of the molecule, thereby shifting the energy levels of the HOMO and LUMO. Such modifications can also alter the oxidation state of the central iron atom, which in turn affects ligand binding affinity and expands the functional diversity of hemoproteins.
Computational Chemistry and Molecular Orbital Analysis
Advances in computational chemistry have become indispensable for visualizing and understanding the complex molecular orbitals of heme. Quantum mechanical methods, particularly density functional theory (DFT), enable detailed simulations of heme’s electronic structure and its interactions with various ligands. These computational models provide insights into the relationships between molecular geometry, electronic properties, and reactivity, deepening our comprehension of heme’s role in biological systems.
Applications in Biomimetic Chemistry
The study of heme’s molecular orbitals extends beyond natural biochemistry, informing the design of synthetic analogs and biomimetic catalysts. Insights into the electronic configurations of heme guide the development of artificial oxygen carriers and catalytic systems, which have potential applications in medicine and industrial chemistry. By mimicking the precise electronic and structural features of heme, chemists aim to create novel materials and therapeutic agents with enhanced bioactivity and efficiency.
Summary and Significance
Exploring the molecular orbitals of heme reveals the intricate interplay between its structural elements and functional capabilities. The coordination of π and σ bonding, the influence of substituents, and the insights gained from computational studies collectively underscore the sophistication of heme’s chemical behavior. This knowledge not only enriches our understanding of fundamental biological processes but also inspires innovative approaches in synthetic chemistry and biotechnology. As research progresses, the detailed study of heme’s molecular orbitals will continue to illuminate the molecular foundations of life and drive advancements in science and technology.
FAQ
What are molecular orbitals of heme?
Molecular orbitals of heme are the combined atomic orbitals from iron and the porphyrin ring atoms, forming bonding and antibonding orbitals that define heme's electronic structure and biological function.
How do molecular orbitals affect heme's function?
They determine how heme binds oxygen and other ligands by influencing electron distribution and orbital interactions, critical for oxygen transport and enzymatic reactions.
What role does iron play in heme's molecular orbitals?
Iron's d orbitals interact with nitrogen p orbitals in the porphyrin ring, creating molecular orbitals essential for oxygen binding and electronic transitions.
How is computational chemistry used to study heme?
Techniques like density functional theory (DFT) simulate heme's electronic structure, helping researchers visualize molecular orbitals and understand reactivity.





Leave a Reply